融点の推定
氷の結晶成長や融解過程をその場観察するとともに、氷の融点を計算機シミュレーションで推定する手順を解説します。
0. 環境設定
各自の PC 上で計算することにします。
0-0 この文書について
以下のように表示されている部分は、特に断りがない限り、Terminal に打ちこむコマンドを表しています。
cd MeltingPoint0-1 自分の PC に Gromacs の動作環境を作る
-
VSCode をインストールする。
- VSCode に以下の拡張をインストールします。

- Dev Container: Docker の仮想マシンにアクセスするのに必要です。
- Git Extension Pack
- gromacs helper: データが見やすくなります。
- VSCode に以下の拡張をインストールします。
-
Docker を用いて、仮想 Linux マシンを自分の PC の中に作ります。
Mac の場合
- Orbstack をインストール https://orbstack.dev/download (Mac 用の Docker Desktop よりもだいぶ便利)
Windows の場合
- Docker Desktop をインストール https://docs.docker.com/desktop/setup/install/windows-install/
-
Linux 仮想マシンを起動します。Terminal をひらき、
docker run -it ubuntuと入力すると、プロンプト”#“が表示されます。続いて、仮想マシン内に、最低限必要なソフトウェアをインストールします。
apt update apt upgrade -y apt install -y python3 pip gromacs git python3-poetry(途中で Time Zone を聞いてくるので Asia/Tokyo を答えて下さい。)
パッケージ名 説明 python3 Python 言語 pip Python のパッケージマネージャ gromacs 分子動力学シミュレーションソフトウェア git バージョン管理ソフトウェア python3-poetry Python 仮想環境 -
ここからの作業は VSCode 内のほうが便利です。教材を展開し、Python の実行環境を作ります。
-
VSCode で新しいウィンドウを開き、Welcome ペインのなかの青い字「Clone Git Repository」をクリックします。VSCode のウィンドウ上方の入力窓で 1. [Clone from GitHub] 2.
vitroid/gromacs-usecasesと入力して Return/Enter を押す 3./root/を選んで OK をクリック 4. 展開した教材をひらくかどうか聞いてくるので、Open を押します。 これで、教材が仮想マシンの/rootフォルダー内に展開されます。ここで使うのは MeltingPoint の中身だけです。 -
VSCode 内でターミナルをひらきます。メニュー →Terminal→New Terminal。
- Terminal 内で、MeltingPoint フォルダーに移動します。
cd MeltingPoint- Python の実行環境を準備します。
poetry install poetry shell
-
0-2 その他のツールの準備
- データをプロットするツールを利用します。Igor Pro や gnuplot や ngraph を使える人はそれを使って下さい。いずれもない場合は Excel や Numbers、それもない場合は Google Spreadsheet などでも代用できます。ただし、個々のプログラムの使い方はここでは解説しません。
- シミュレーション結果を可視化するツールVMDをあらかじめインストールしておいて下さい。
1. 水分子モデルを選ぶ
分子モデルとは、分子の形や相互作用を簡単な関数で近似したものです。本来、分子間に働く力や、結合のしかたなどは電子状態が関わっており、電子の軌道(原子核の周囲に雲のように分布する)を正確に計算してはじめて得られるものですが、分子シミュレーションでは計算量を減らすために、結合長を固定したり、電子分布を点電荷で近似したりします。
計算機シミュレーションに利用される水分子モデルは 100 種類以上あります。近似を行う際のコンセプトが違うためです。そのうちのほとんどは、常温の液体の水や水溶液の性質が再現されるように設計されてきたため、結晶化相転移を観察するのに適していません。(融点が 273 K からかなりずれています)
ここでは、近年開発され、信頼性の高い TIP4P/Ice モデルを利用します。このモデルの 1 気圧での融点は 270 K と言われており[^1]、常温以下での水や氷やハイドレート(水和結晶)のふるまいを研究するのによく使われています。
氷の融点なんて、実験ですでにわかっているだろうって?その通りです。でも、コンピュータシミュレーションで用いる水分子モデルの融点が知りたいのです。モデルの質が悪ければ、融点は実験値とはあわないでしょう。分子シミュレーションの目的は、実験と合うデータを得ることではありません。実験を再現することを保証しつつ、実験では見えない情報をとりだすことに価値があります。融点などの物性をよく制限するモデルであれば、分子の運動もそれなりに正しく再現していると考えられます。そうであれば、例えば固液界面からどのように水が凍っていくのかをその場で観察した結果(これは実験では見えない)をも信頼できると期待できます。
TIP4P/Ice の形状の詳細はリンク先を見て下さい。
水分子は 3 原子でできているのですが、相互作用を質点間の相互作用で近似的に表現するために、もう一点相互作用点を追加してあるのが四点モデルと呼ばれる水モデルの特徴です。
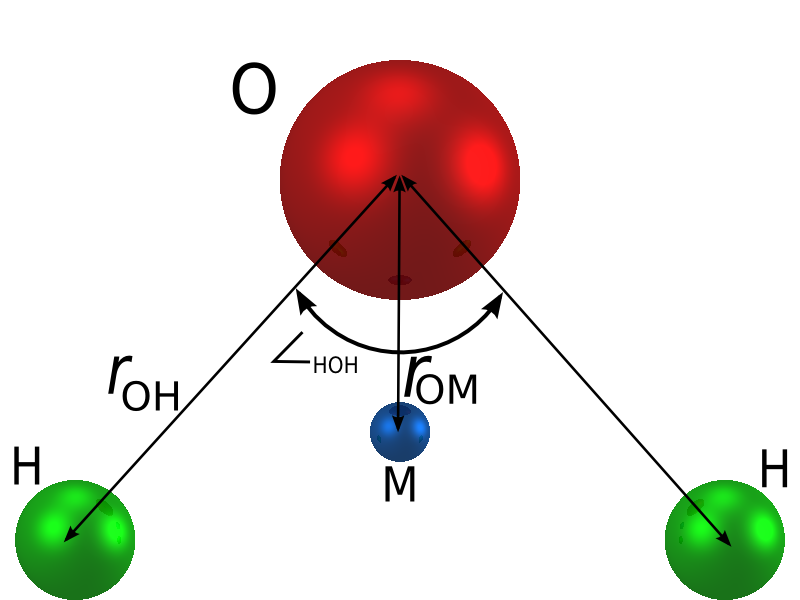
TIP4P/Ice の分子モデルはice.topに書かれています。
; taken from http://www.sklogwiki.org/SklogWiki/index.php/GROMACS_topology_file_for_the_TIP4P/Ice_model
[ defaults ]
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ
1 2 no 1.0 1.0
[atomtypes]
;name mass charge ptype sigma epsilon
IW 0 0.000 D 0.0 0.0
OWT4 15.99940 0.000 A 0.31668 0.88211
HW 1.00800 0.000 A 0.00000E+00 0.00000E+00
...全部で 50 行ぐらいのデータファイルで、ここには以下のような内容が書かれています。
- 1 つの水分子をいくつの点で近似表現しているか。
- 分子の中での点の相対距離
- 点と点はどのように相互作用するか(Coulomb 力と Lennard-Jones 力)
- 分子は点と点をバネでつないだ柔軟な物体として扱うのか、それとも剛直で変形しない剛体として扱うのか。
TIP4P/Iceの場合は
- 相互作用点は 4 点。
- H-H 間距離は約 0.15 nm、O-H 間距離は約 0.1 nm。
- 3 つが Coulomb 相互作用し、残る 1 点は Lennard-Jones 相互作用。
- 剛体として扱う。
となっています。
2. 分子を配置する
最初に、分子をどう配置するかは、あとのシミュレーションの成否に大きく関わります。
融点を推定する際に、例えば初期配置として、すべての分子が氷の結晶構造に並ぶ配置を選ぶと、融点より 10 K や 20 K 温度を上げても氷は融けません(過熱現象)。これは、融けはじめるきっかけとなる、構造の崩壊が偶然起こるまでの時間がかかりすぎるためです。もちろん、無限に近い計算時間があれば、融点より上では融けはじめるはずですが、そんなに長い計算は今のコンピュータでは不可能です。
同じように、はじめに完全に液体の状態からはじめると、温度をいくらさげても自然に結晶が生じるまでには非常に時間がかかります。(過冷却現象)
そこで、融点を推定したい場合には、あらかじめ氷と水が共存する状態を準備します。こうすれば、設定した温度が融点よりも高ければ、氷は徐々に融けていきますし、低ければ氷の成長が見られます。
初期配置は、次の手順で準備します。
- GenIceツールを使い、氷の結晶構造を作ります。結晶は 1:1:2 の長い直方体形状になるようにします。
- 水分子の半分を「固定」します。文字通り、最初の場所から動けなくします。
- 温度を 800 K まで上げて、短い分子動力学シミュレーションを行います。固定した部分は構造を保ちますが、それ以外の水分子はすぐに融解します。
- 温度を戻し、固定を解除します。これにより、液体と固体が半分ずつの初期構造ができます。
- 固定を解除した状態で、270 K(融点)付近でしばらくシミュレーションを行い、構造を緩和させます。
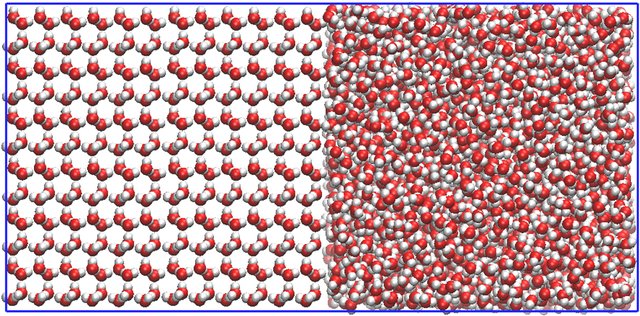
右半分が融けた氷。左半分は固定されていて動かない。[^3]周期境界条件なので、左端と右端、上端と下端、手前と奥はそれぞれつながっていて分子が出入りできる。
以下に、具体的な手順を書きます。
2-1 氷の結晶を作る
GenIceは、さまざまな氷の結晶構造を生成するツールです。これを用い、氷 Ih(こおりいちえいち、六方晶氷 I、もっとも身近に存在する氷)を生成します。
まず、ターミナルを開いてください。
genice2 で氷の構造を生成します。
genice2 1h -r 3 3 6 -w tip4p > ice.gro-rオプションで、単位胞を x,y,z 方向にいくつならべるかを指定しています。また、-wで水分子モデルの種類を指定します。”>“は画面に表示される内容をファイルに書きだすことを表す記法です。
ice.groの末尾には、こんな風に箱の大きさが nm 単位で書かれています。
...
863ICE HW1 3450 2.178 1.983 5.379
863ICE HW2 3451 2.220 2.107 5.303
863ICE MW 3452 2.217 2.020 5.310
864ICE OW 3453 1.694 1.648 5.310
864ICE HW1 3454 1.740 1.620 5.231
864ICE HW2 3455 1.697 1.744 5.306
864ICE MW 3456 1.700 1.657 5.300
2.34685163 2.20607179 5.42191111x, y, z 軸方向の寸法がそれぞれ 2.3 nm, 2.2 nm, 5.4 nm で、z 方向に長いセルです。
とりあえず、この構造を可視化してみましょう。VSCode のウィンドウ左側のファイルブラウザでice.groを右クリックし、ダウンロードします。そのファイルを VMD で開きます。
2-2 分子を固定する
Gromacsには、原子の番号を指定して、その原子を動けなくする機能があります。手で指定するのはたいへんなので、python スクリプトをつかって、Z 座標が 2.7 nm 未満の原子だけを固定します。
2-2-1 原子にインデックスをつける
Gromacs の make_ndx コマンドは、原子を種類ごとに分類した.ndxファイルを生成します。
gmx make_ndx -f ice.gro -o ice.ndxを実行し、prompt が表示されたら、q を押して終了します。ice.ndx には以下のような内容が書かれます。
[ System ]
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
16 17 18 19 20 21 22 23 24 25 26 27 28 29 30
31 32 33 34 35 36 37 38 39 40 41 42 43 44 45
...このファイルには、原子がグループ分けされています。System グループには、全原子が属しています。
同じ要領で、固定したい原子だけを列挙した FIX グループを新たに自分で準備しましょう。
2-2-2 固定した原子のインデックスを追加する
このファイルの末尾に、固定したい原子のインデックスを追加します。(”>>“は既存のファイルの後ろに書きたすことを表す記法です)
python3 zfix.py 0.0 2.7 < ice.gro >> ice.ndxこれにより、ice.ndxファイルの末尾に、以下のような行が追加されます。
...
[FIX]
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40
41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
...2-3 残り半分を融かす
体積一定のままで、温度を 800 K まで上げます。普通なら沸騰しそうなものですが、膨張できないので、液体になるだけでとまります。このステップは、ただ構造を壊したいだけなので、長時間実行する必要はありません。
Gromacs での分子動力学法の実行は、いつも 3 段階の手順になります。
- 設定ファイルを「コンパイル」して、Gromacs が読みやすいデータ形式に変換する。
- そのデータを読みこんで、分子動力学シミュレーションの本体(mdrun)を実行する。
- 生成したデータを、人間が読みやすいように変換する。
2-3-1 コンパイル
拡張子.mdpのファイルには、分子動力学の実施方法を詳細に記載します。個々のパラメータの説明は、fix.mdpファイルにコメントとして書きこんであります。計算時間、記録間隔、温度設定、圧力設定など、分子配置と相互作用以外のほぼすべての情報は.mdpに書きます。fix.mdpの場合、それらに加えて.ndxファイルで指定したFIX原子グループを固定することを指示します。
以下のコマンドでこれをコンパイルして、.tprファイルを作ります。
gmx grompp -maxwarn 1 -f fix.mdp -p ice.top -c ice.gro -n ice.ndx -o fixed.tprこれにより、Gromacs が読みこむファイルfixed.tprができました。
2-3-2 実行
2-3-1 で作った 00001.tpr を読みこんでシミュレーションを実行します!
gmx mdrun -deffnm fixedこれを実行すると、ほかにもfixed.からはじまるたくさんのファイルが生成されます。(-deffnm は、たぶん default file name の略で、明示的に指定しなかった出力ファイルには全部fixed.をつけて下さい、と指示しています。)
fixed.cpt: チェックポイントファイル、続きの計算を行うのに必要なファイル。fixed.edr: 温度や圧力などの、統計情報が含まれるファイル。fixed.trr: 原子の座標や速度のデータ(トラジェクトリと呼ばれる)。fixed.log: その他の記録。実行時間などもこのファイルに記録される。
fixed.logの末尾には以下のように実行時間の情報が記録されます。
starting mdrun 'water TIP4P/Ice'
50000 steps, 100.0 ps.
Writing final coordinates.
Core t (s) Wall t (s) (%)
Time: 792.815 99.102 800.0
(ns/day) (hour/ns)
Performance: 87.185 0.275これは MacBook Air で実行したケースですが、
- CPU を 800%使っている(8 core を使用している)
- 50000 ステップ実行した。1 ステップが 0.002 ps なので、100 ps のシミュレーションが行われた。
- かかった時間は 100 秒。このペースだと一日で 90 ns 分の計算ができる。
といったことが読みとれます。
2-3-3 確認
シミュレーション結果を可視化するツールVMDを使い、分子運動を可視化してみましょう。
まず、分子動力学計算で得られた分子配置を、可視化しやすいように変換します。
gmx trjconv -f fixed.trr -s fixed.tpr -pbc whole -o fixed.gro入力を求められたら、0 を押してリターンを押します。(System 全体を可視化します)
これで、fixed.groファイルができました。かなり大きなファイルです。
- VSCode で、
fixed.groを右クリックしてダウンロードします。 - VMD を起動します。
- Download したファイルを、VMD の Main ウィンドウにドラッグアンドドロップします。
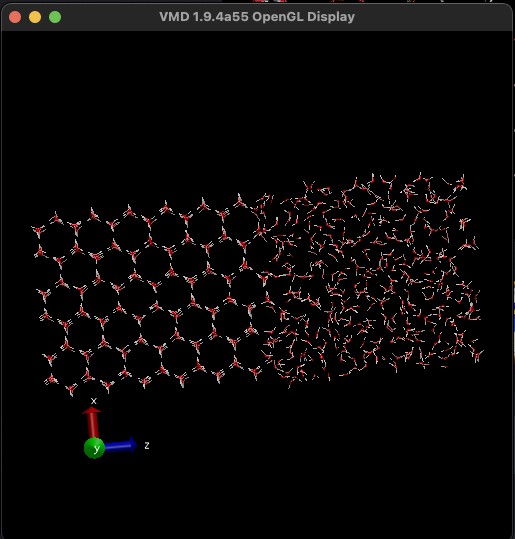
固定した部分は、分子はびくとも動いていないことがわかります。一方、固定していない部分は完全にランダムになっています。
2-3-4 緩和
温度を下げ、原子の固定をはずして、構造を緩和させます。この時に、体積も変化できるようにし、圧力一定でのシミュレーションに切り替えます。
シミュレーションの設定はさきほどと同じく、.mdpに書きます。今回はrelax.mdpを使用します。
VSCode のファイル比較機能を使うと、relax.mdpとfix.mdpの違いがわかります。
変更されているのは 3 点。
- 温度が 270 K に下げられた。
- 体積一定から圧力一定に切り替えた。
- 分子の固定を外した。
これを使って、またコンパイルし、実行します。
gmx grompp -maxwarn 1 -f relax.mdp -p ice.top -c fixed.tpr -t fixed.cpt -o relaxed.tpr
gmx mdrun -deffnm relaxed2-3-3 と同じように、relaxed.groを作って、VMD で観察して下さい。今度は、氷の部分の分子もぷるぷると振動していることがわかります。
gmx trjconv -f relaxed.trr -s relaxed.tpr -pbc whole -o relaxed.gro3. 目標とする温度で、シミュレーションを行う
2.で作った構造を初期配置とし、いろんな温度で長時間のシミュレーションを行います。
圧力は 1 気圧、シミュレーション時間は 3〜10 ns を目標とします。このシミュレーションでは dt=0.002 ps としていますので、1 ns は 500 000 ステップに相当します。
ここでは例として、250 K での計算を解説します。これは、TIP4P/Ice の融点[^1]に対して 20 K も低温なので、氷はただちに成長すると予想されます。(250 K 以外の温度の場合は適宜読みかえて下さいね。)
3-1 作業環境の準備
継続計算用の設定ファイルcontinue.mdpを VSCode で開き、下に示すように温度設定を 250 K に修正して、ファイル名はT250.mdpとして別名で保存します。
...
ref_t = xxxx ; 系の温度。単位はK。
...また、T250.mdpのはじめの方に、計算ステップ数の指示があります。低温と高温では必要なステップ数が違います。とりあえず、280 K 以上では 3 ns、それ未満では 10 ns のシミュレーションを行いたいのですが、講義時間内に計算が終わらない可能性を考え、まず全員 3 ns を計算して下さい。(あとから延長できます)
10 ns の計算に挑戦する場合は、以下の部分を書きかえて下さい。
...
nsteps = 5000000 ; MDのステップ数。
nstxout = 5000 ; 座標がnstxoutステップに一度出力される。
nstlog = 5000 ; logに情報が書き込まれる頻度。
...relax.mdpとcontinue.mdpを比較して下さい。
3-2 シミュレーションの実行
そして、設定ファイルをコンパイルします。生成するファイル名はT250.tprとしました。
gmx grompp -maxwarn 1 -f T250.mdp -p ice.top -c relaxed.tpr -t relaxed.cpt -o T250.tpr実行!
gmx mdrun -deffnm T250さっきの 30 倍〜100 倍の時間がかかるはずです。コーヒーでも飲んで、気長に待ちましょう。
3-3 延長
あとの解析で、まだシミュレーションの時間が足りないことがわかった場合は、次のようにして計算を延長することができます。
まず、さきほどgromppで生成した.tprファイルをもとに、実行時間を延長した新しい.tprファイルを作成します。 -extendオプションは追加計算する時間を ps 単位で指定します。2000 ps=1 ns だけ延長します。
gmx convert-tpr -extend 2000 -s T250.tpr -o extended.tprそして実行! (継続計算の場合は、オプションをいろいろ指定する必要があるようです。)
gmx mdrun -deffnm extended -cpi T250.cpt -s extended.tpr -noappendこんどは食事ができるぐらい時間がかかるかもしれません。
4. 結果の可視化
実際にどんなことが起こっているのかを確認するために、分子座標データを手許のコンピュータに転送し、VMD で分子運動を表示させてみましょう。
まず、トラジェクトリデータ.trrを、.gro形式に変換します。
(シミュレーションの途中でも変換できます。その場合は、ターミナルをもう一つ開いて作業します。)
gmx trjconv -f T250.trr -s T250.tpr -pbc whole -o T250.groこれを Download し、VMD で開きます。
1 ns の計算では、融解しつつあるのか凍結しつつあるのか判別が難しい場合がありますが、10 ns 計算すれば、おおよそ判別がつきます。
温度を下げればより速く凍るか、というとそうでもなくて、温度が低いと分子運動が遅くなるせいで、結晶成長も遅くなります。
温度が高ければ高いほどはやく融けるのは間違いありません。
5. 判定
可視化しても成長の様子が目に見えてわかるようになるまでには、かなり時間を要することがわかります。そこで、違うアプローチをとります。
圧力と温度一定でシミュレーションした場合、液体になるか固体になるかで密度が違ってきます。同時に、ポテンシャルエネルギーも違います。固体のほうが必ず(エントロピーが低いので)ポテンシャルエネルギーも低くなるはずです。つまり、ポテンシャルエネルギーか密度の時間変化を見れば、氷が成長しつつあるのか、融解しつつあるのかを見分けることができます。
これをプロットして確認します。
シミュレーション中の温度や圧力、ポテンシャルエネルギーなどの熱力学量の時間変化は.logファイルにも書かれますが、より詳しい情報が.edrファイルに記録されます。
この.edrファイルの内容を、人間が読みやすい形に変換するのが、gmx dumpコマンドです。
gmx dump -e T250.edrですが、こうして変換されたファイルも、あとのデータ処理にはあまり便利ではありませんそこで、これをさらに自作スクリプトundump.pyを使って、表形式に変換します。
gmx dump -e T250.edr | python3 undump.py > T250.txtこのファイルをダウンロードし、てきとうなツール(Excel?)を使って開いて、時間とポテンシャルエネルギーの関係をプロットして下さい。
5-1 Gnuplot の場合
第 1 カラムが時刻、第 3 カラムがポテンシャルエネルギー、第 7 カラムが密度です。
plot 'T250.txt' u 1:3 w l6. 融点の推定
十分長いシミュレーションを行えば、融点より高温では氷は完全に融解し、低温では完全に凍結するはずです。そして、ポテンシャルエネルギーや密度は一定値になります。
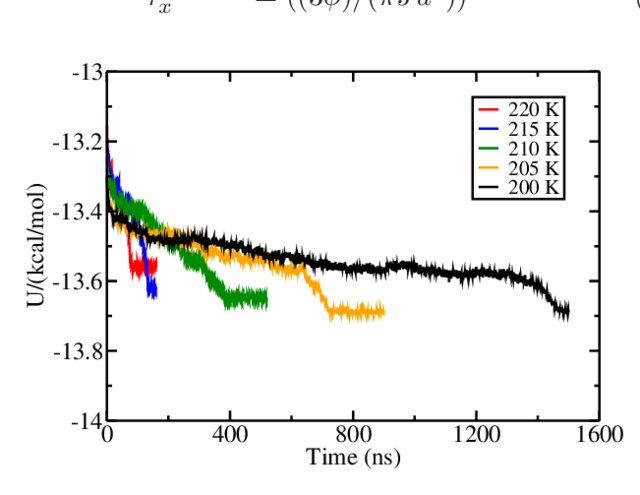
共存状態から氷が成長する過程でのポテンシャルエネルギーの変化。低温ではより低いエネルギーに到達するものの、時間がかかる。 [^4] ここで使われている水分子モデル TIP4P/Ice の氷の融点は約 270 K と推定されており、これらの温度は融点よりもかなり低温に相当する。
一定値になるまでの時間は、融点から遠いほど短くなるはずです(ただし、上にも書いたように、温度がとても低くなると、分子運動が遅くなるために凍るまでにまた時間がかかるようになります。)
仮に、完全凍結/完全融解までの時間が、融点からの温度差に反比例するものとします。その場合は、温度を横軸にとり、縦軸にポテンシャルエネルギーの変化が完了するまでの時間の逆数をとれば、低温側、高温側それぞれに曲線を描け、それらは融点で 0 に漸近する(すなわち、融点丁度では、いくらまっても凍りも融けもしない)と思われます。
しかし、融点に近づいてくると、完全に凍るまでの時間がかかりすぎます。また、十分に長い計算ができない場合も、完全な凍結を確認できません。そんな場合は、最終的なポテンシャルエネルギーが得られるのを待つ代わりに、ポテンシャルエネルギーの減少/増加速度を測定し、その逆数を縦軸にとる、という方法もあります。
7. 課題
全員で手分けして、250 K から 290 K まで(5 K 間隔で、ただし 270 K は除く。)のポテンシャルエネルギーの時間変化を採取し、それらの結果から、TIP4P/Ice の融点を推定して下さい。
(黒板に温度を書き、そこに名前を書いてもらう)
下の論文[^1]によれば、TIP4P/Ice の融点は 270 K となっていますが、今回は高速化のために、分子数がかなり少ない系でシミュレーションを行っています。そのせいで、融点が 270 K からずれる可能性があります。
予想される融点である 270 K や、それに近い温度では、いつまでたっても凍りも融けもしない可能性があります。
相変化が終わるまでにかかる時間は、いつも一定ではありません。時間があれば、同じ温度で何回か測定し、平均値を求めるべきです。
レポートには、以下の情報を書いて下さい。
- 各温度で融解または凍結にかかった時間 (表形式)と その根拠となるグラフ(生データ)。自分の計算した温度以外のデータは、交換・共有して下さい。(余力があれば、自分でほかの温度を計算しても構いません。)
- 温度に対し、所要時間の逆数をプロットしたグラフ。(おおよそでいい)
- 2 のグラフの形から、どのようにして融点を推定したかの説明、考察など。
8. 蛇足
- 最初に作る氷を氷 III や氷 V にし、適切な圧力を指定すれば、高圧氷の融点も推定可能です。
- 高圧では、共存状態から意図しない結晶構造が生じる可能性もあります。ただし、それを見分けるのは難しいでしょう。
- 固体と気体、あるいは液体と気体の間の相転移でも、同じ方法が使えます。
- 固体と固体の間の相転移の場合は、シミュレーションセルの直方体の大きさをどちらかの固体の結晶の単位胞にあわせると、もう一方の結晶にとっては不都合になってしまうため、この方法で転移温度を決めることはできません。その場合は、固有振動数計算から自由エネルギーを求める別の技術を用います。
- 水の場合、液体と、もう一つの液相の間の相転移も観察できます![^2]
- この利用例では初期構造に氷 Ih を選び、半分を融かして固液共存状態を準備しました。これを、圧力だけ高圧に切りかえても、氷 III や氷 V と水の共存状態のシミュレーションにはなりません。結晶が自然に別の結晶に転移するには非常に時間がかかります。氷 Ih に、氷 V ほどの高圧を加えると、構造が崩壊して全部液体になるでしょう。
- terminal を複数開き、フォルダーをコピーして、いくつもの条件で同時に計算することもできます。
8. あと始末
レポートも書きおわって、データが要らなくなったら、Orbstack/Docker Desktop で Image(OS のインストール CD に相当)と Container(展開後のハードディスクイメージ)の両方を消すとディスク使用量を減らせます。
References
[^1] Conde, M. M., Rovere, M. & Gallo, P. High precision determination of the melting points of water TIP4P/2005 and water TIP4P/Ice models by the direct coexistence technique. J. Chem. Phys. 147, 244506 (2017).
[^2] Yagasaki, T., Matsumoto, M. & Tanaka, H. Spontaneous liquid-liquid phase separation of water. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 89, 020301 (2014).
[^3] Yeyue Xiong, Parviz Seifpanahi Shabane, and Alexey V. Onufriev*, Melting Points of OPC and OPC3 Water Models, ACS Omega 39, 25087–25094 (2020).
[^4] Espinosa, J. R., Sanz, E., Valeriani, C. & Vega, C. Homogeneous ice nucleation evaluated for several water models. J. Chem. Phys. 141, 18C529 (2014).
2024 年度は使わない
0-1 クラウドを利用するための準備
- 計算サーバ(岡山大学、あるいは Amazon EC2)にログインするためのアカウントをこちらから配布します。岡山大学の計算機を利用する場合は、まず VPN 接続の準備をし、接続できることを確認して下さい。(→9. 補遺)
- VSCode のウィンドウ左のアイコンから、以下の拡張を追加して下さい。
- Remote SSH
- 所定の IP アドレスに接続します。VSCode のウィンドウ左下のボタン
 を押して下さい。(いろいろ聞いてきます。)
を押して下さい。(いろいろ聞いてきます。) - クラウドのコンピュータにプログラムソースを準備します。VSCode のウィンドウ中央の
Clone Git Repository...を押し、Clone from GitHubと表示されたらリターンを押し、vitroid/gromacs-usecasesを指定して下さい。保存先を聞かれたらリターンを押します。2 回目のログイン以降はこの作業は不要です。 - ソースを保存したフォルダーの、
MeltingPointフォルダーを開きます。