氷表面の観察
氷を成長させて、氷の表面(気固界面)を観察します。
0. 環境設定
0-0 この文書について
以下のように表示されている部分は、特に断りがない限り、Terminalに打ちこむコマンドを表しています。
cd MeltingPoint0-1 実行環境の準備
-
コードをダウンロードし、計算サーバのいずれかで展開します。(緑のボタンを押すと、Download ZIPボタンが表示されます。)
計算サーバ上の空き要領のあるディスク上でZIPファイルを展開しで下さい。
0-2 手許のコンピュータの準備
- 手許のパソコンに、VSCodeをインストールし、VSCodeから計算サーバにリモートアクセスして、IceGrowthフォルダーを開いて下さい。
- シミュレーション結果を可視化するツールVMDをあらかじめインストールしておいて下さい。
1. 水分子モデルを選ぶ
計算機シミュレーションに利用される水分子モデルには非常に多数の種類があります。そのうちのほとんどは、常温の液体の水や水溶液の性質が再現されるように設計されてきたため、結晶化相転移を観察するのに適していません。
ここでは、近年開発され、信頼性の高いTIP4P/Iceモデルを利用します。このモデルの1気圧での融点は270 Kと言われており[^1]、常温以下での水や氷やハイドレート(水和結晶)のふるまいを研究するのによく使われています。
氷の融点なんて、実験ですでにわかっているだろうって?その通りです。でも、コンピュータシミュレーションで用いる水分子モデルの融点が知りたいのです。モデルの質が悪ければ、融点は実験値とはあわないでしょう。分子シミュレーションの目的は、実験と合うデータを得ることではありません。実験を再現することを保証しつつ、実験では見えない情報をとりだすことに価値があります。融点などの物性をよく制限するモデルであれば、分子の運動もそれなりに正しく再現していると考えられます。そうであれば、例えば固液界面からどのように水が凍っていくのかをその場で観察した結果(これは実験では見えない)をも信頼できると期待できます。
TIP4P/Iceの形状の詳細はリンク先を見て下さい。
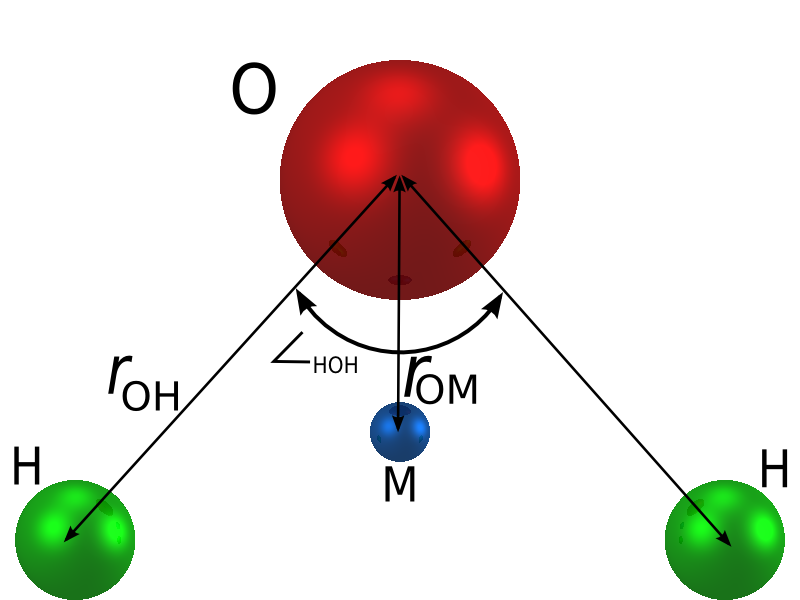
水分子は3原子でできているのですが、相互作用を質点間の相互作用で近似的に表現するために、もう一点相互作用点を追加してあるのが四点モデルと呼ばれる水モデルの特徴です。
TIP4P/Iceの分子モデルはice.topに書かれています。
; taken from http://www.sklogwiki.org/SklogWiki/index.php/GROMACS_topology_file_for_the_TIP4P/Ice_model
[ defaults ]
; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ
1 2 no 1.0 1.0
[atomtypes]
;name mass charge ptype sigma epsilon
IW 0 0.000 D 0.0 0.0
OWT4 15.99940 0.000 A 0.31668 0.88211
HW 1.00800 0.000 A 0.00000E+00 0.00000E+00
...全部で50行ぐらいのデータファイルで、ここには以下のような内容が書かれています。
- 1つの水分子をいくつの点で近似表現しているか。
- 分子の中での点の相対距離
- 点と点はどのように相互作用するか(Coulomb力とLennard-Jones力)
- 分子は点と点をバネでつないだ柔軟な物体として扱うのか、それとも剛直で変形しない剛体として扱うのか。
TIP4P/Iceの場合は
- 相互作用点は4点。
- H-H間距離は約0.15 nm、O-H間距離は約0.1 nm。
- 3つがCoulomb相互作用し、残る1点はLennard-Jones相互作用。
- 剛体として扱う。
となっています。
2. 分子を配置する
最初に、分子をどう配置するかは、あとのシミュレーションの成否に大きく関わります。
融点を推定する際に、例えば初期配置として、すべての分子が氷の結晶構造に並ぶ配置を選ぶと、融点より10 Kや20 K温度を上げても氷は融けません(過熱現象)。これは、融けはじめるきっかけとなる、構造の崩壊が偶然起こるまでの時間がかかりすぎるためです。もちろん、無限に近い計算時間があれば、融点より上では融けはじめるはずですが、そんなに長い計算は今のコンピュータでは不可能です。
同じように、はじめに完全に液体の状態からはじめると、温度をいくらさげても自然に結晶が生じるまでには非常に時間がかかります。(過冷却現象)
そこで、融点を推定したい場合には、あらかじめ氷と水が共存する状態を準備します。こうすれば、設定した温度が融点よりも高ければ、氷は徐々に融けていきますし、低ければ氷の成長が見られます。
初期配置は、次の手順で準備します。
- GenIceツールを使い、氷の結晶構造を作ります。結晶は1:1:2の長い直方体形状になるようにします。
- 水分子の半分を「固定」します。文字通り、最初の場所から動けなくします。
- 温度を800 Kまで上げて、短い分子動力学シミュレーションを行います。固定した部分は構造を保ちますが、それ以外の水分子はすぐに融解します。
- 温度を戻し、固定を解除します。これにより、液体と固体が半分ずつの初期構造ができます。
- 固定を解除した状態で、270 K(融点)付近でしばらくシミュレーションを行い、構造を緩和させます。
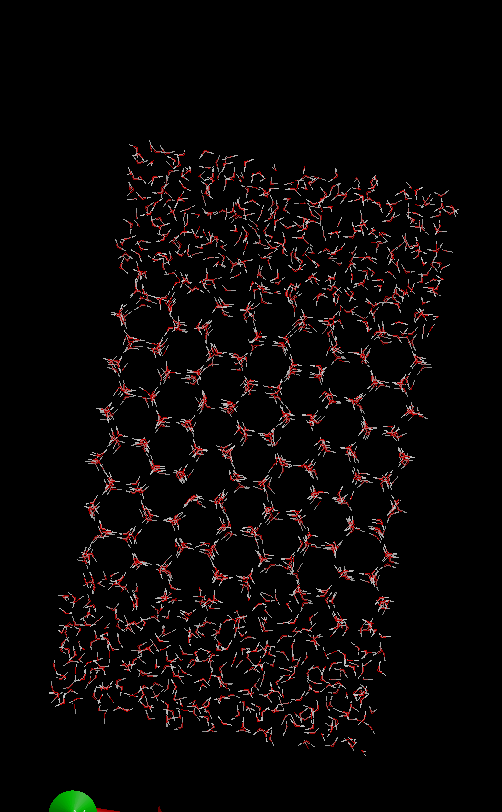
半分が融けた氷。中央の氷部分は固定されていて動かない。周期境界条件なので、左端と右端、上端と下端、手前と奥はそれぞれつながっていて分子が出入りできる。
以下に、具体的な手順を書きます。
2-1 氷の結晶を作る
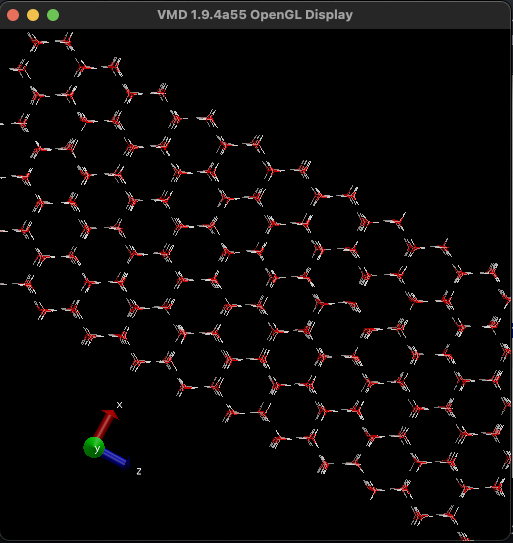
GenIceは、さまざまな氷の結晶構造を生成するツールです。これを用い、氷Ih(こおりいちえいち、六方晶氷I、もっとも身近に存在する氷)を生成します。
ターミナルで以下のように入力します。
genice2 1h -r 4 4 6 -w tip4p > ice.gro-rオプションで、単位胞をx,y,z方向にいくつならべるかを指定しています。また、-wで水分子モデルの種類を指定します。
ice.groの末尾には、こんな風に箱の大きさがnm単位で書かれています。
...
1535ICE HW1 6138 3.090 2.720 5.304
1535ICE HW2 6139 3.000 2.842 5.305
1535ICE MW 6140 3.010 2.755 5.304
1536ICE OW 6141 2.482 2.391 5.301
1536ICE HW1 6142 2.523 2.357 5.380
1536ICE HW2 6143 2.393 2.357 5.304
1536ICE MW 6144 2.476 2.382 5.312
3.12913551 2.94142906 5.42191111x, y, z軸方向の寸法がそれぞれ3.1 nm, 2.9 nm, 5.4 nmで、z方向に少し長いセルです。
とりあえず、この構造を可視化してみましょう。VSCodeのウィンドウ左側のファイルブラウザでice.groを右クリックし、ダウンロードします。そのファイルをVMDで開きます。
2-2 分子を固定する
Gromacsには、原子の番号を指定して、その原子を動けなくする機能があります。手で指定するのはたいへんなので、pythonスクリプトをつかって、Z座標が2.7 nm未満の原子だけを固定します。
2-2-1 原子にインデックスをつける
Gromacsのmake_ndxコマンドは、原子を種類ごとに分類した.ndxファイルを生成します。
gmx make_ndx -f ice.gro -o ice.ndxを実行し、promptが表示されたら、qを押して終了します。ice.ndxには以下のような内容が書かれます。
[ System ]
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
16 17 18 19 20 21 22 23 24 25 26 27 28 29 30
31 32 33 34 35 36 37 38 39 40 41 42 43 44 45
...このファイルには、原子がグループ分けされています。Systemグループには、全原子が属しています。
同じ要領で、固定したい原子だけを列挙したFIXグループを新たに自分で準備しましょう。
2-2-2 固定した原子のインデックスを追加する
このファイルの末尾に、固定したい原子のインデックスを追加します。
python3 zfix.py 2.0 3.4 < ice.gro >> ice.ndxこれにより、ice.ndxファイルの末尾に、以下のような行が追加されます。
...
[FIX]
2049 2050 2051 2052 2057 2058 2059 2060 2061 2062 2063 2064 2065
2066 2067 2068 2069 2070 2071 2072 2077 2078 2079 2080 2081 2082
2083 2084 2085 2086 2087 2088 2097 2098 2099 2100 2101 2102 2103
2104 2105 2106 2107 2108 2109 2110 2111 2112 2113 2114 2115 2116
2121 2122 2123 2124 2125 2126 2127 2128 2129 2130 2131 2132 2133
2134 2135 2136 2141 2142 2143 2144 2145 2146 2147 2148 2149 2150
...2-3 残り半分を融かす
体積一定のままで、温度を800 Kまで上げます。普通なら沸騰しそうなものですが、膨張できないので、液体になるだけでとまります。このステップは、ただ構造を壊したいだけなので、長時間実行する必要はありません。
Gromacsでの分子動力学法の実行は、いつも3段階の手順になります。
- 設定ファイルを「コンパイル」して、Gromacsが読みやすいデータ形式に変換する。
- そのデータを読みこんで、分子動力学シミュレーションの本体(mdrun)を実行する。
- 生成したデータを、人間が読みやすいように変換する。
2-3-1 コンパイル
拡張子.mdpには、分子動力学の実施方法を詳細に記載します。個々のパラメータの説明は、fix.mdpファイルにコメントとして書きこんであります。計算時間、記録間隔、温度設定、圧力設定など、分子配置と相互作用以外のほぼすべての情報は.mdpに書きます。fix.mdpの場合、それらに加えて.ndxファイルで指定したFIX原子グループを固定することを指示します。
以下のコマンドでこれをコンパイルして、.tprファイルを作ります。
gmx grompp -maxwarn 1 -f fix.mdp -p ice.top -c ice.gro -n ice.ndx -o fixed.tprこれにより、Gromacsが読みこむファイルfixed.tprができました。
2-3-2 実行
2-3-1で作った00001.tprを読みこんでシミュレーションを実行します!
gmx mdrun -deffnm fixedこれを実行すると、ほかにもfixed.からはじまるたくさんのファイルが生成されます。(-deffnmは、たぶんdefault file nameの略で、明示的に指定しなかった出力ファイルには全部fixed.をつけて下さい、と指示しています。)
fixed.cpt: チェックポイントファイル、続きの計算を行うのに必要なファイル。fixed.edr: 温度や圧力などの、統計情報が含まれるファイル。fixed.trr: 原子の座標や速度のデータ(トラジェクトリと呼ばれる)。fixed.log: その他の記録。実行時間などもこのファイルに記録される。
fixed.logの末尾には以下のように実行時間の情報が記録されます。
starting mdrun 'water TIP4P/Ice'
50000 steps, 100.0 ps.
Writing final coordinates.
Core t (s) Wall t (s) (%)
Time: 792.815 99.102 800.0
(ns/day) (hour/ns)
Performance: 87.185 0.275これはMacBook Airで実行したケースですが、
- CPUを800%使っている(8 coreを使用している)
- 50000ステップ実行した。1ステップが0.002 psなので、100 psのシミュレーションが行われた。
- かかった時間は100秒。このペースだと一日で90 ns分の計算ができる。
といったことが読みとれます。
2-3-3 確認
シミュレーション結果を可視化するツールVMDを使い、分子運動を可視化してみましょう。
まず、分子動力学計算で得られた分子配置を、可視化しやすいように変換します。
gmx trjconv -f fixed.trr -s fixed.tpr -pbc whole -o fixed-snapshots.gro入力を求められたら、0を押してリターンを押します。(System全体を可視化します)
これで、fixed-snapshots.groファイルができました。かなり大きなファイルです。
- VSCodeで、
fixed-snapshots.groを右クリックしてダウンロードします。 - VMDを起動します。
- Downloadしたファイルを、VMDのMainウィンドウにドラッグアンドドロップします。
固定した部分は、分子はびくとも動いていないことがわかります。一方、固定していない部分は完全にランダムになっています。
)
2-3-4 緩和
温度を下げ、原子の固定をはずして、構造を緩和させます。この時に、体積も変化できるようにし、圧力一定でのシミュレーションに切り替えます。
シミュレーションの設定はさきほどと同じく、.mdpに書きます。今回はrelax.mdpを使用します。
VSCodeのファイル比較機能を使うと、relax.mdpとfix.mdpの違いがわかります。
変更されているのは3点。
- 温度が270 Kに下げられた。
- 体積一定から圧力一定に切り替えた。
- 分子の固定を外した。
これを使って、またコンパイルし、実行します。
gmx grompp -maxwarn 1 -f relax.mdp -p ice.top -c fixed.tpr -t fixed.cpt -o relaxed.tpr
gmx mdrun -deffnm relaxed2-3-3と同じように、relaxed.groを作って、VMDで観察して下さい。今度は、氷の部分の分子もぷるぷると振動していることがわかります。
gmx trjconv -f relaxed.trr -s relaxed.tpr -pbc whole -o relaxed-snapshots.gro2.5 気相を追加
セルを伸長し、気相の領域を準備します。
relaxed.groをコピーしてstretch.groという名前で保存します。
stretch.gro末尾のセルサイズのz方向のサイズを、2倍ていど(だいたいでいい)に伸ばします。
1536water OW 6141 1.862 2.746 5.038 0.2622 0.0469 0.2826
1536water HW1 6142 1.818 2.718 5.118 1.7875 -0.9183 0.8374
1536water HW2 6143 1.835 2.837 5.026 0.6920 0.5086 2.4287
1536water MW 6144 1.853 2.755 5.047 0.5253 -0.0208 0.6461
3.14308 2.95454 10そして、再度構造緩和させますが、今回は体積を変化させないようにします。この設定はstretch.mdpに書いてあります。
VSCodeのファイル比較機能で、relax.mdpとstretch.mdpを比べて下さい。
またコンパイルし、実行します。
gmx grompp -maxwarn 1 -f stretch.mdp -p ice.top -c stretch.gro -o stretched.tpr
gmx mdrun -deffnm stretchedちゃんと三相共存で安定化していることをVMDで確認します。
gmx trjconv -f stretched.trr -s stretched.tpr -pbc whole -o stretched.gro3. 目標とする温度で、シミュレーションを行う
2.5で作った構造を初期配置とし、いろんな温度で長時間のシミュレーションを行います。
圧力は1気圧、シミュレーション時間は3〜10 nsを目標とします。このシミュレーションではdt=0.002 psとしていますので、1 nsは500 000ステップに相当します。
ここでは例として、250 Kでの計算を解説します。これは、TIP4P/Iceの融点[^1]に対して20 Kも低温なので、氷はただちに成長すると予想されます。(250 K以外の温度の場合は適宜読みかえて下さいね。)
3-1 作業環境の準備
継続計算用の設定ファイルcontinue.mdpをVSCodeで開き、下に示すように温度設定を250 Kに修正して、ファイル名はT250.mdpとして別名で保存します。
...
ref_t = xxxx ; 系の温度。単位はK。
...また、T250.mdpのはじめの方に、計算ステップ数の指示があります。低温と高温では必要なステップ数が違います。とりあえず、280 K以上では3 ns、それ未満では10 nsのシミュレーションを行いますので、適宜その部分も書きかえて下さい。
...
nsteps = 5000000 ; MDのステップ数。
nstxout = 5000 ; 座標がnstxoutステップに一度出力される。
nstlog = 5000 ; logに情報が書き込まれる頻度。
...relax.mdpとcontinue.mdpを比較して下さい。
3-2 シミュレーションの実行
そして、設定ファイルをコンパイルします。生成するファイル名はT250.tprとしました。
gmx grompp -maxwarn 1 -f T250.mdp -p ice.top -c relaxed.tpr -t relaxed.cpt -o T250.tpr実行!
gmx mdrun -deffnm T250 -nt 8並列処理の数-nt 8を指定しています。(つけないと途中でエラーを出すケースがありました。でも要らないかも。) 実習では32 coreの計算機を利用するので、最大32まで指定できますが、みんなで同時に実行する可能性があるので、混んでいる時は8または16にとどめて下さい。
さっきの30倍〜100倍の時間がかかるはずです。コーヒーでも飲んで、気長に待ちましょう。
3-3 延長
あとの解析で、まだシミュレーションの時間が足りないことがわかった場合は、次のようにして計算を延長することができます。
まず、さきほどgrompppで生成した.tprファイルをもとに、実行時間を延長した新しい.tprファイルを作成します。 -extendオプションは追加計算する時間をps単位で指定します。2000 ps=1 nsだけ延長します。
gmx convert-tpr -extend 2000 -s T250.tpr -o extended.tprそして実行! (継続計算の場合は、オプションをいろいろ指定する必要があるようです。)
gmx mdrun -deffnm extended -cpi T250.cpt -s extended.tpr -noappendこんどは食事ができるぐらい時間がかかるかもしれません。
4. 結果の可視化
実際にどんなことが起こっているのかを確認するために、分子座標データを手許のコンピュータに転送し、VMDで分子運動を表示させてみましょう。
まず、トラジェクトリデータ.trrを、.gro形式に変換します。
gmx trjconv -f T250.trr -s T250.tpr -pbc whole -o T250-snapshots.groこれをDownloadし、VMDで開きます。
1 nsの計算では、融解しつつあるのか凍結しつつあるのか判別が難しい場合がありますが、10 ns計算すれば、おおよそ判別がつきます。
温度を下げればより速く凍るか、というとそうでもなくて、温度が低いと分子運動が遅くなるせいで、結晶成長も遅くなります。速く凍る温度を見付けるために、いくつかの温度で試して下さい。
5. 解析
5.1 横断水素結合数
完全にアイスルールを満足する、分極のない氷であれば、どのような断面で切っても、その面を横切る入出結合本数は等しくなるはずです。
水素結合数のバランスを確認します。
hbbalance.pyは、シミュレーションセルをz軸方向に0.01 nm単位でスライスし、各スライス面を横切る水素結合の本数を数えます。
python3 hbbalance.py < T250.groを実行すると、.groに含まれているフレーム数分のファイル(拡張子.hbb.txt)が生成されます。中身は以下のような感じです。
0.0 -2.0 1.0 3.0
0.01 -4.0 9.0 13.0
0.02 -4.0 12.0 16.0
0.03 -3.0 20.0 23.0
0.04 -3.0 25.0 28.0
0.05 -4.0 28.0 32.0
0.06 -2.0 32.0 34.0
0.07 -3.0 35.0 38.0
0.08 -2.0 41.0 43.0
...第1カラムがスライス面のz座標、第3カラムが面を正方向に横切る水素結合の本数、第4カラムが負方向に横切る水素結合の本数、第2カラムがそれらの差です。
アイスルールが満足され、全分極が0の氷であれば、この値はどこの面で切っても必ず0になるはずです。逆に言えば、この指標が0でなければ、アイスルールが満足されていないか、全分極が非0であることを意味します。
5.2 環位相統計
成長して生じた氷が、均質な水素無秩序氷と言えるかどうかを、環位相統計で調べます。
cycles.pyは、水素結合ネットワークのなかにあるすべての既約六員環(より小さい環の組みあわせで表現できない6員環)を、水素結合の向きで分類し個数を数えます。
python3 cycles.py < T250.groを実行すると、.groに含まれているフレーム数分のファイル(拡張子.cyc.txt)が生成されます。中身は以下のような感じです。
0 276 291.94520547945206
1 423 437.917808219178
3 456 437.917808219178
5 115 109.4794520547945
7 215 218.958904109589
9 55 54.73972602739725
11 121 109.4794520547945
21 4 4.561643835616438第1カラムが位相、第2は.groファイル内での出現頻度、第3カラムは、第二カラムと同じ環総数の場合の理想分布。
初期配置では液体領域があり、その中にも六員環が存在するので、この総数は液体の六員環の数も含んでいます。GenIceで生成した氷では、種類別の環の個数は理想分布(別紙)に一致し、液体の場合も理想分布から外れる理由がないので、初期配置での環種分布は理想分布にほぼ一致すると思われます。一方、氷が成長する間に、偏った種類の環が多めに生じるなら、結晶成長につれてこの分布は理想分布からずれてくるでしょう。
複数の.cyc.txtファイルを並べて、位相3の環の個数だけを列挙する方法。例えば、以下のスクリプトで、10〜10000.cyc.txt(10個おき)を順番にならべ、位相3の情報だけを抽出できます。
seq 1 1000 | sed -e 's/$/0.cyc.txt/' | xargs cat | grep '^3 ' > cyclestat.3.txt分解して説明します。
seq 1 1000は、1〜1000の数列を標準出力します。sed -e 's/$/0.cyc.txt/'は、標準入力の行末($)を、0.cyc.txtに置きかえ標準出力します。xargs catは、標準入力の内容をcatコマンドの引数とみなして実行します。これで、10〜10000.cyc.txtが順番に並んだ大きなテキストファイルが標準出力されます。grep '^3 'は、標準入力の内容のうち、行頭が3ではじまる行だけを抽出します。
この結果をgnuplotで見れば、環数の時間変化がよみとれます。
6. 成長面のいれかえ
GenIceで生成したIh構造は、z軸方向がプリズム面$(1010)$ですらなく、$(11\bar 20)$方向となっているようだ。成長方向をベーサル面$(0001)$またはプリズム面にするためには、z軸とxまたはy軸を交換する必要がある。
GenIceには、単位胞の向きを変える機能がある。例えば、x軸とz軸を交換する操作は、行列で書けば、次のようになる。 $$\left(\begin{matrix}0& 0&1\0&1&0\1&0&0 \end{matrix}\right)$$ 同様に、y軸とz軸の交換は次のようになる。 $$\left(\begin{matrix}1& 0&0\0&0&1\0&1&0 \end{matrix}\right)$$
GenIceには、結晶構造の情報を読みこみ、変形させて新たな結晶構造プラグインを生成する機能がある。→https://github.com/vitroid/GenIce/wiki/Transform-the-unit-cell
新たに生成したプラグインをlatticesに格納しておくと、それを使って変形した結晶構造を生成できる。
mkdir lattices
genice2 1h -f 'reshape[0,0,1,0,1,0,1,0,0]' > lattices/1hprism.py
genice2 1h -f 'reshape[1,0,0,0,0,1,0,1,0]' > lattices/1hbasal.pyで、新たな結晶構造プラグインを作り、これを使って結晶を生成する。
genice2 1hbasal -r 4 4 6 -w tip4p > basal.gro
genice2 1hprism -r 4 4 6 -w tip4p > prism.gro確認のため、VMDで表示してみよう。basal.groは、z軸方向から見ると正六角形の配列が見えるはずだ。
9. 補遺
9-1 VPN接続
岡山大学理論化学研究室所有の計算サーバ(Xeon 96 core, IP 192.168.3.220)にリモートアクセスします。設定情報は別途お渡しします。
うまく接続できない場合は、Amazon EC2を利用します。
9-1-1 Macの場合
System Settings->VPN->Add VPN Configurationで、設定を入力します。
https://www.cc.uec.ac.jp/ug/ja/remote/vpn/l2tp/macos114/index.html が参考になります(入力する内容は適宜おきかえて下さい)
9-1-2 Windowsの場合
https://www.seil.jp/saw-mpc/doc/sa/pppac/use/pppac-client/win11_l2tp.html が参考になります(入力する内容は適宜おきかえて下さい)
コントロールパネル→ネットワークと共有センター→アダプタの設定変更→VPNを選ぶ→プロパティ→セキュリティ→次のプロトコルを許可する→CHAPにチェック!!! (ふう。なんでこんなに深いの)
9-2 Amazon EC2の個人利用
Amazon EC2の無料枠でも、そこそこの計算はできます。
- AWSにアカウントを作ります。クレジットカード番号が必要になります。
- EC2ダッシュボードで、EC2インスタンスを作成します。インスタンスタイプt2.micro (2 cores)までなら、無料で利用できます。
- OSにはUbuntuを選びます。(計算プラットホームとして利用するのに適しています)
- そのほか、ほとんどの設定はデフォルトのままでいいですが、不明な点は講師にお尋ね下さい。
9-3 Gromacsとその他のツールのインストール
クラウドを使わなくても、Unix系のOSになら、GromacsやGenIceを簡単にインストールできます。
9-3-1 Ubuntu/Debian Linuxの場合
(管理者権限で実行する必要があります。)
apt update
apt install gromacs python3 python3-pip
pip install numpy
pip install genice29-3-2 Redhat/Amazon Linux/CentOS7の場合
EC2のAmazon Linux/RedHat Linuxではgromacsパッケージが見付かりませんでした。RPMから必要なものを個別にインストールする必要があるようです。
9-3-3 MacOSの場合
Homebrewをセットアップしておきましょう。
HomeBrewでは、管理者権限なしにインストールできます。
brew install gromacs python3
pip install genice29-3-4 Windowsの場合
(情報求む!)
References
[^1] Conde, M. M., Rovere, M. & Gallo, P. High precision determination of the melting points of water TIP4P/2005 and water TIP4P/Ice models by the direct coexistence technique. J. Chem. Phys. 147, 244506 (2017).
[^2] Yagasaki, T., Matsumoto, M. & Tanaka, H. Spontaneous liquid-liquid phase separation of water. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 89, 020301 (2014).
[^3] Yeyue Xiong, Parviz Seifpanahi Shabane, and Alexey V. Onufriev*, Melting Points of OPC and OPC3 Water Models, ACS Omega 39, 25087–25094 (2020).
[^4] Espinosa, J. R., Sanz, E., Valeriani, C. & Vega, C. Homogeneous ice nucleation evaluated for several water models. J. Chem. Phys. 141, 18C529 (2014).